Accurate assignment of amino-acid side chains remains a major bottleneck in macromolecular structure determination, particularly for low-resolution structures, samples derived from natural sources, and proteins exhibiting sequence heterogeneity. Even when global validation metrics are satisfactory, local side-chain ambiguities can propagate errors into functional interpretation and computational downstream analyses. The SEQUENCE SLIDER framework provides a foundation for automated sequence assignment by integrating structural biology data and phylogenetic analysis (PMID: 32133987; PMID: 35104880). However, local implementation and large-scale calculations limit accessibility for many experimental biologists.
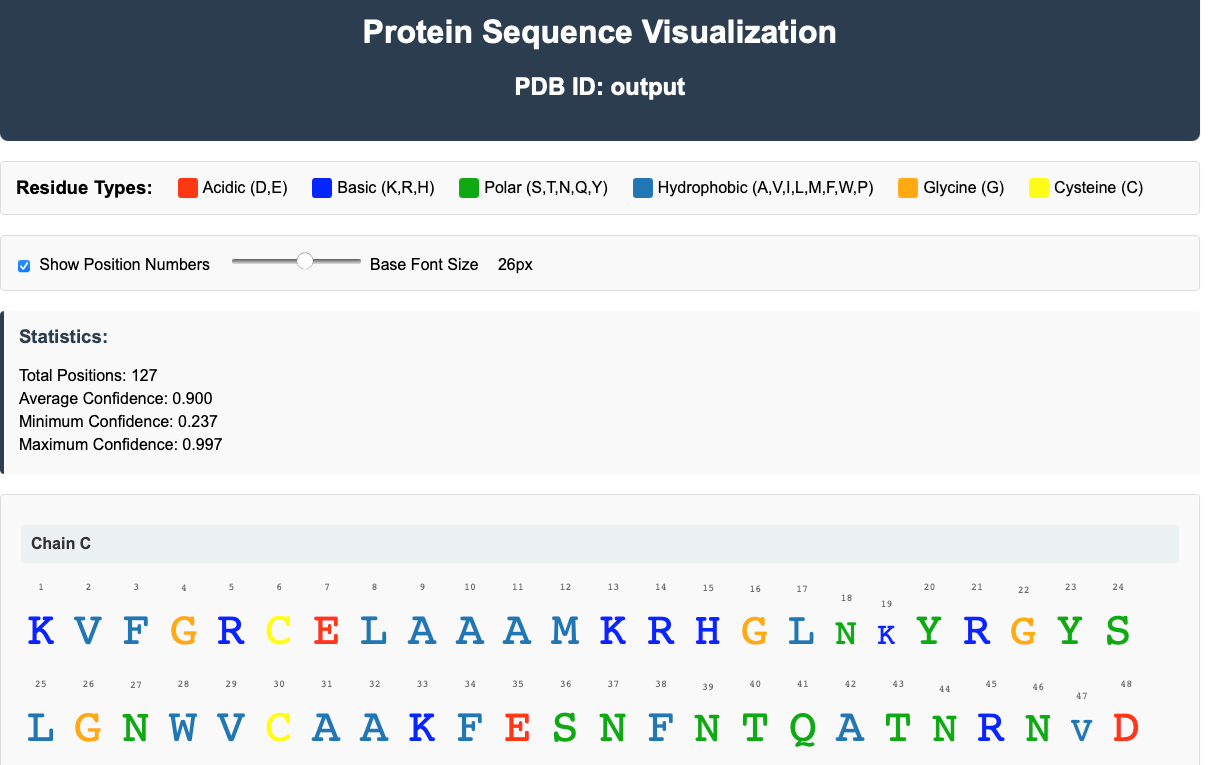
SEQUENCE SLIDER-ML is an interactive web server that enables users to evaluate, rank, and visualize residue-specific side-chain hypotheses directly from structural data. Users upload a coordinate file (PDB or mmCIF) together with an experimental electron density map. The server analyzes the local structural environment and ranks alternative amino-acid hypotheses for each residue using an internal machine-learning–assisted confidence scoring model.
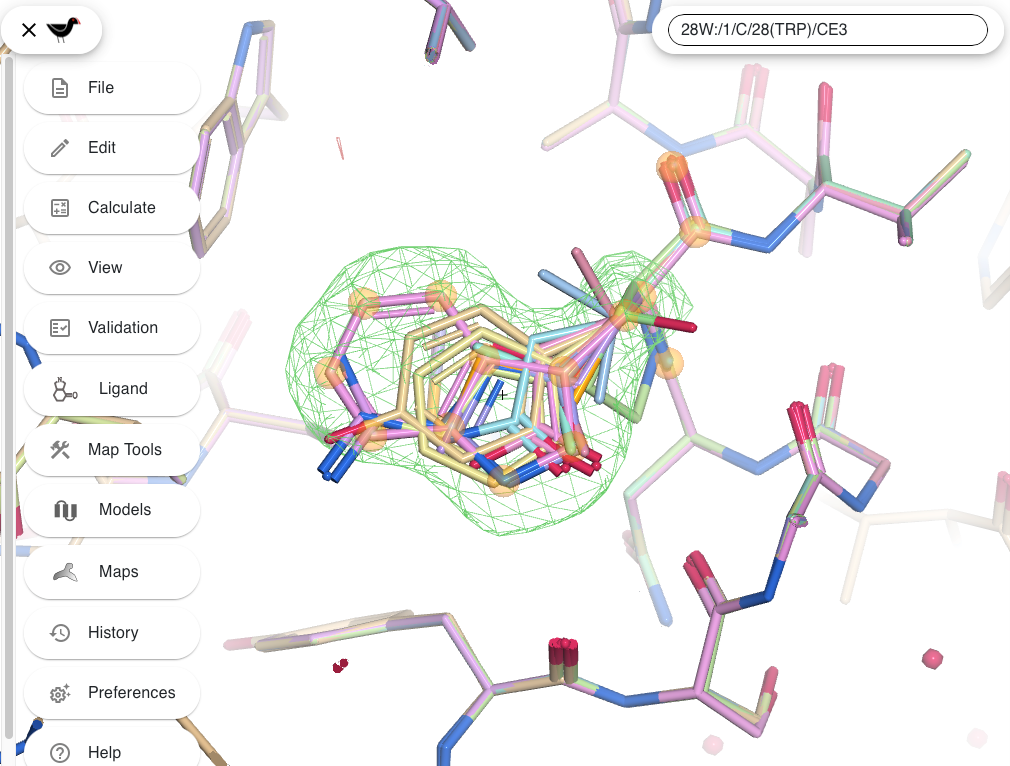
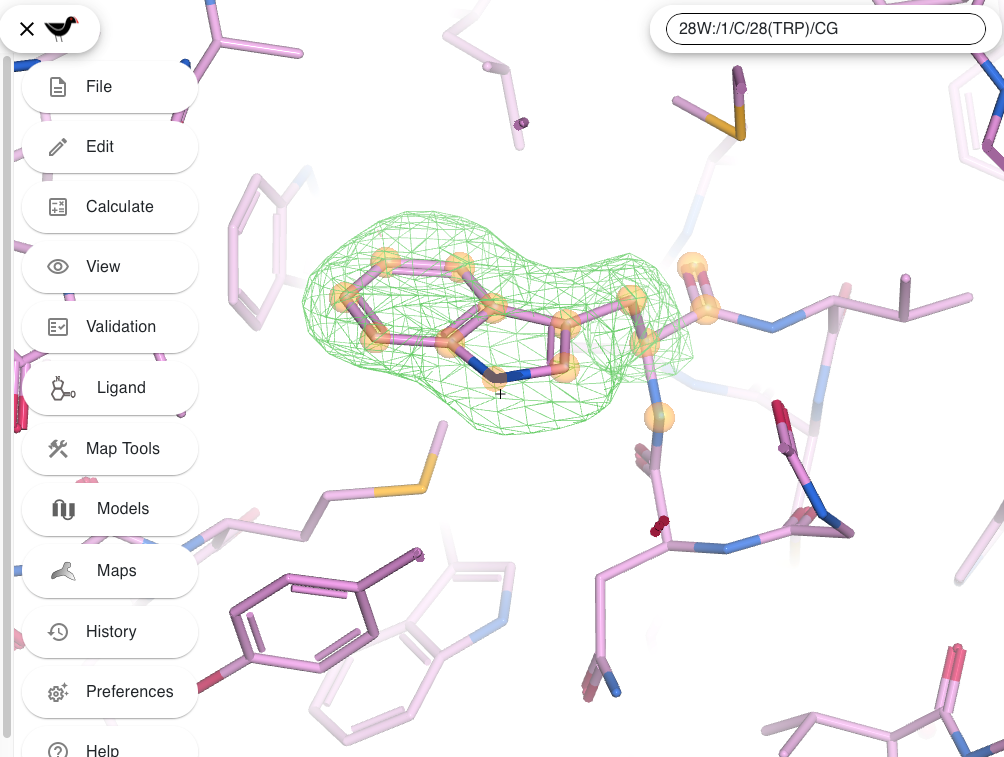
Results are presented through interactive sequence-level and 3D visualizations (with moorhen and GitHub repository), allowing users to inspect alternative hypotheses in the context of the experimental map. The server highlights low-confidence cases that require expert judgment, providing objective decision support rather than automated assignments.
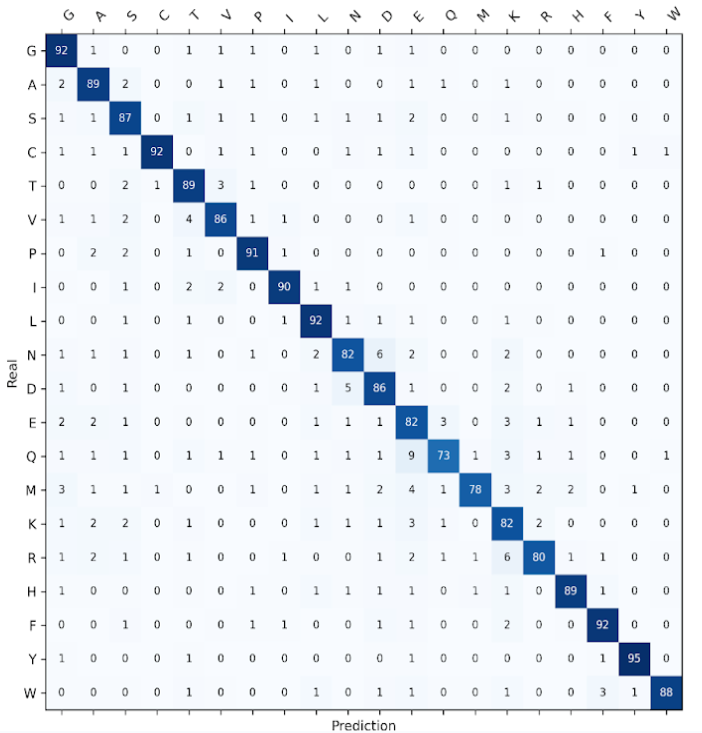
The machine-learning component is used exclusively as an internal scoring engine. It is trained on curated experimentally solved protein structures and validated using blind, independent test sets, with model interpretability provided through SHAP analysis.
The server complements existing model-building and refinement software and is particularly useful for ambiguous regions, heterogeneous samples, and post-refinement validation.